
ここからヘッダーメニューです。



ここからサイト内検索です。
ここから共通メニューです。 ここまで共通メニューです。
自分の意志に反して手足、顔面をピクつかせたり動かしてしまう舞踏運動と認知機能障害、精神症状(幻覚、妄想、抑うつなど)をきたす遺伝性、進行性の神経疾患です。常染色体優性遺伝ですので、性別に関係なく遺伝子異常が50%の確率で子孫に伝わります。大脳基底核の一部である尾状核に存在する小型神経細胞の脱落により、GABA作動性抑制神経細胞が機能低下をきたすことで舞踏運動を生じると考えられています。遺伝子の異常として、第4染色体短腕上に局在しているHTT遺伝子のDNA配列3個(シトニン・アデニン・グアニン)の繰り返しが異常に増加します(GACリピート)。DNA配列3つの異常伸長が原因となる遺伝性疾患は神経疾患に多くトリプレットリピート病と呼び、ハンチントン病もこれに含まれます。本邦での有病率は10万人あたり1人弱程度とされ欧米より少なく、神経変性疾患の中では稀な疾患です。
発症年齢は30~50歳が多いとされますが、高齢で発症することもあります。発症は緩徐で、初期症状としては不随意運動、性格変化、認知症が高頻度に出現します。具体的には、細かい運動がしにくくなった、よく物を落とすなどといった巧緻運動障害、意図に反して顔をしかめたり手先が勝手に動いてしまうといった不随意運動、また落ち着きがない、怒りっぽくなった、無気力、抑うつ状態などといった多彩な精神症状を認めます。これらの症状は緊張時や興奮時に増悪、睡眠中には消失する特徴があります。
症状が進行すると不随意運動や運動持続困難のため、歩行が不安定になり転倒しやすくなり、また呂律が回らなくなったり嚥下が困難になります。徐々に社会生活が営めなくなり、末期には重度の認知症をきたしほぼ寝たきりの状態に至ります。
優性遺伝の形式をとるため、両親のどちらかが同じ病気であることがほとんどですが、一般に子供のほうが若い年齢で発症し重症化する傾向があり(表現促進現象)、この傾向は男親が病気である場合により顕著にみられます。
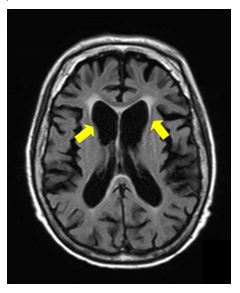
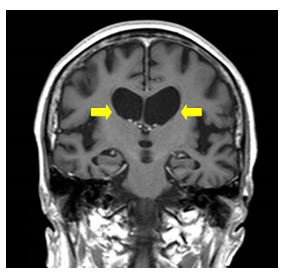
血液検査や脳脊髄液検査では疾患に特異的な異常はみられません。頭部CT・MRIにて大脳基底核の一部である尾状核と呼ばれる部位の萎縮がおこり、進行とともに全脳が委縮、それに伴い側脳室前角の拡大を認め、診断の一助となります。脳血流シンチグラムでは前頭・側頭葉型の血流低下を認めます。下に示す写真はハンチントン病のMRI画像です(黄の矢印で示した尾状核の萎縮のほか、大脳皮質の広範な萎縮もみられます)。
|
|
現在のところ根本的に治す、あるいは進行を遅らせる治療法は確立されていませんが、対症療法としてお薬を用いることで症状をある程度コントロールすることが可能です。
舞踏運動に対しては、ドパミン受容体拮抗薬(抗精神病薬)が有効ですが、舞踏運動の程度と副作用のバランスを見極めつつ薬剤の種類や量を調整することが重要です。また攻撃性や抑うつなどの精神症状に対する治療薬も考慮します。ハンチントン病の保険適用のあるテトラベナジンは、抑うつ症状を引き起こし、悪化させるリスクが高いため当院では採用していません。
主に精神症状によると考えられますが、家系・家族内で孤立を来す傾向があり、自殺により亡くなられる方も多く報告されています。また一緒に生活あるいは介護されるご家族の負担も大変大きなものがあります。そのため、社会資源の有効活用や他科との連携により、包括的な診療を行っていく必要があります。当院では、外来での経過観察、加療を行いつつ、年に1度程度の定期的な入院により、病状評価やリハビリを行い、療養環境の調整も併せて行うようにしております。特定疾患(いわゆる難病)の医療費助成制度や介護保険制度を利用することが重要になります。
逓信病院Webサイトのご利用について | 外来診療のご案内 | 入院・面会のご案内 | 人間ドック | 診療科のご案内 | 病院のご紹介
![]()
![]()
![]()